High Precision Protein Function Prediction with Graph Contrastive Representation Learning
Abstract
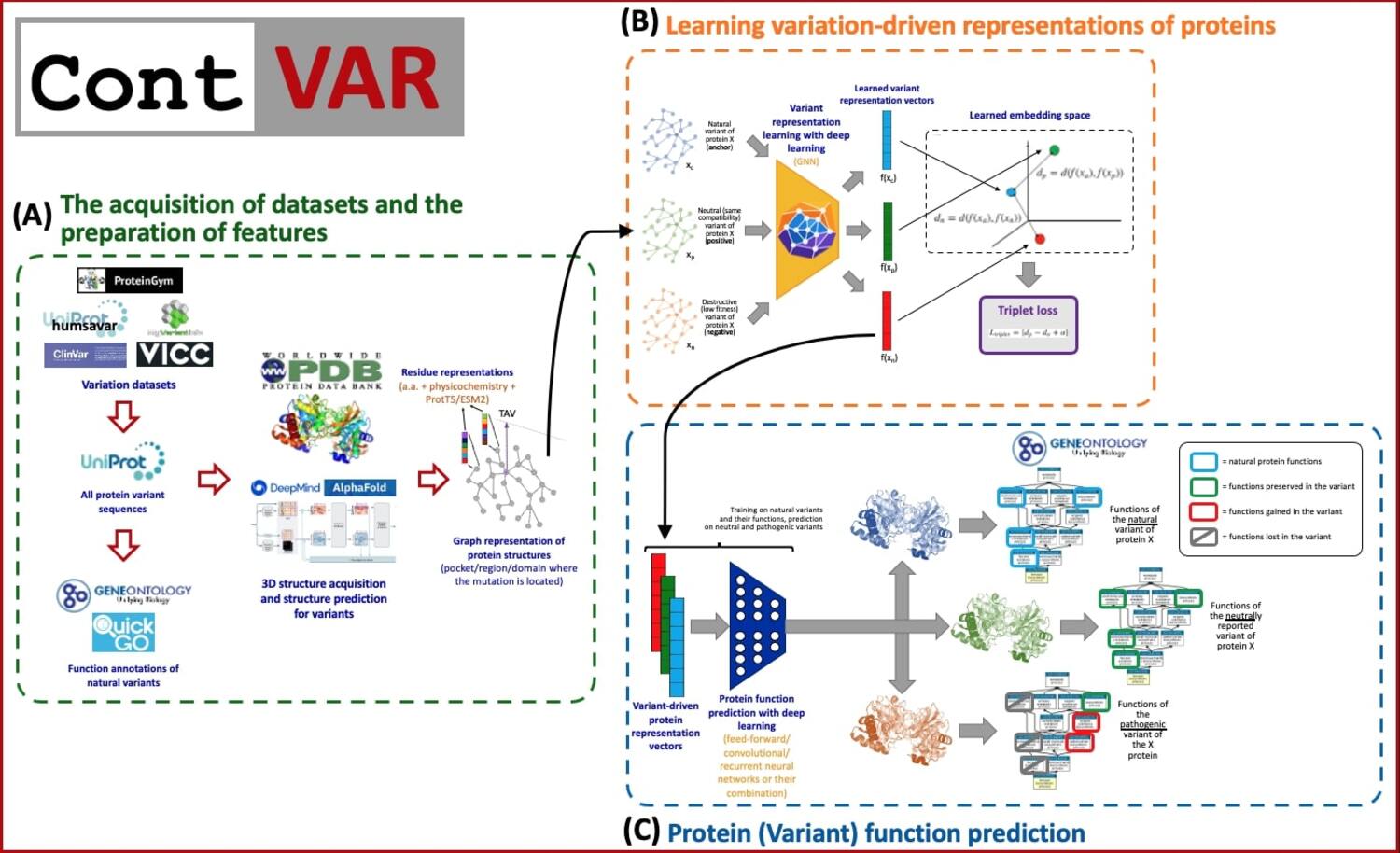
This project addresses the challenge of understanding functional alterations in proteins caused by single amino acid variations, a key issue in biomedical research and biotechnology. Existing computational methods often indicate whether a mutation is pathogenic or leads to a broad gain or loss of function but cannot specify the exact biological roles affected. To overcome this, we introduce ContVAR, an artificial intelligence–based framework that combines contrastive and graph representation learning. Its core is a self-supervised contrastive variant representation learning model that captures detailed structural and functional information from protein variants without relying on extensive annotations. Due to the lack of large-scale gained/lost function datasets, deep mutational screening data support this self-supervised approach, enabling the model to distinguish between function-altering and neutral variants. Using 3D structures predicted by AlphaFold2/3, encoded as graphs, ContVAR produces high-dimensional embeddings that feed into a supervised model trained on Gene Ontology annotations of natural proteins. The system will be validated with resources such as UniProt and ProteinGym and examined in biologically important contexts like the p53-MDM2 pathway, with additional verification through cell-based assays in liver, breast, and colon cancer models. Finally, ContVAR will be released as a user-friendly computational tool with a graphical interface, providing life science researchers with a practical means to analyze protein variants in real time and accelerate applications in drug discovery and biotechnology.