Disease Centric Large Scale De Novo Design of Drug Candidate Molecules with Graph Generative Deep Adversarial Networks
Abstract
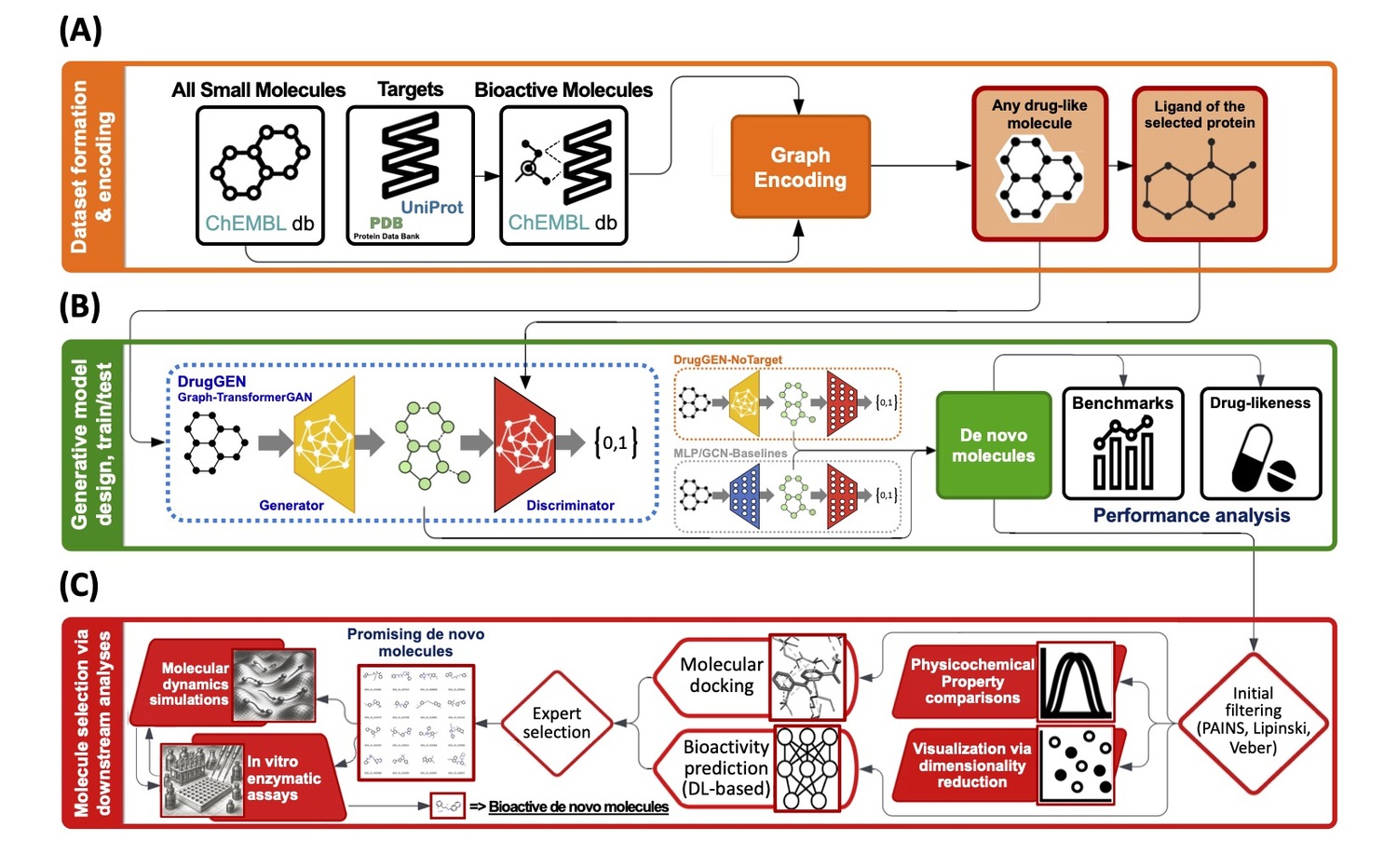
Discovering novel, efficacious small molecules remains a slow and failure-prone process, given the enormity of chemical space. We developed DrugGEN, a generative AI system for de novo, target-specific small-molecule design against drug-resistant cancers. Molecules were represented as graphs and modelled with a graph-transformer-based generative adversarial network conditioned on target context. The model ingested curated training corpora and was optimised to maximise chemical validity, uniqueness/novelty, and predicted bioactivity while satisfying multi-parameter property profiles. In Stage 1, we trained and evaluated the system on expert-selected cancer targets, demonstrating high validity and novelty rates, with improved predicted affinity compared to strong baselines. A prioritised subset of designs was advanced to medicinal chemistry; multiple compounds were synthesised successfully and exhibited measurable inhibitory activity on enzymatic assays and drug-resistant cancer cell lines in vitro. In Stage 2, prospective rounds incorporated feedback from docking, molecular dynamics, and assay results, yielding iterative gains in predicted potency and developability. We released an open-access codebase (https://github.com/HUBioDataLab/druggen) and a web service (https://huggingface.co/spaces/HUBioDataLab/DrugGEN) that enables life-science researchers to generate candidate molecular structures by specifying target proteins, facilitating rapid hypothesis generation and experimental triage. DrugGEN establishes a practical, target-conditioned molecular generation pipeline that couples graph-native modelling with property-aware constraints to deliver experimentally testable inhibitors for resistant oncology indications.